
This page compiles our content related to acid-base and electrolyte disorders. For further information on diagnosis and treatment, follow the links below to our full BMJ Best Practice topics on the relevant conditions and symptoms.
Introduction
Relevant conditions
Evaluation of respiratory acidosis | go to our full topic on Evaluation of respiratory acidosis Respiratory acidosis occurs when arterial partial pressure levels of carbon dioxide (PCO₂) increase above the normal range of 35 to 45 mmHg, due to inefficient clearance of CO₂. This leads to an accumulation of hydrogen ions, causing the arterial pH to fall below 7.35. It may be acute or chronic, and failure to recognize and treat the underlying cause can lead to respiratory failure and death. Causes of respiratory acidosis include COPD, multilobar pneumonia, foreign body aspiration, drug use (such as sedatives, anesthetics, alcohol, narcotics), and oxygen therapy in patients with COPD. Chronic respiratory acidosis is commonly caused by obesity and COPD. Clinical features of respiratory acidosis include respiratory depression (hypoventilation), obtundation, hemodynamic instability, and respiratory muscle fatigue (accessory muscle use, dyspnea, tachypnea). |
|---|---|
Evaluation of respiratory alkalosis | go to our full topic on Evaluation of respiratory alkalosis Respiratory alkalosis is an acid-base disorder characterized by a primary reduction in the arterial PCO₂ below the normal range of 35 to 45 mmHg, leading to an increase in pH above 7.45 and a subsequent decrease in bicarbonate from a normal value of 24 mEq/L. The decrease in PCO₂ typically occurs as a result of alveolar hyperventilation with an excess of CO₂ excretion compared to production.[3] The etiologies of respiratory alkalosis are multiple and include pulmonary embolism, sepsis and systemic inflammatory response syndrome, acute respiratory distress syndrome, pneumonia, and hyperventilation syndrome.[3] Respiratory alkalosis can be acute or chronic in nature. |
Evaluation of metabolic acidosis | go to our full topic on Evaluation of metabolic acidosis Metabolic acidosis is indicated by an arterial pH of less than 7.35, a decrease in the plasma bicarbonate level, and/or a marked increase in the anion gap (calculated by subtracting the sum of major measured anions, chloride and bicarbonate, from the major measured cation, sodium). Where the anion gap is normal (6-12 mEq/L), gastrointestinal or renal causes are common.[4] This is also referred to as hyperchloremic or non-anion gap metabolic acidosis. Where the anion gap is increased, causes include diabetic ketoacidosis, alcoholic ketoacidosis, lactic acidosis, kidney disease, or ingestion of methanol, ethanol, ethylene glycol, propylene glycol, 5-oxoproline (e.g., in patients with chronic ingestion of acetaminophen), or salicylic acid. With simple metabolic acidosis, the normal adaptive respiratory response will decrease the arterial PCO₂ 1.0 to 1.5 times the decrease in serum hydrogen carbonate (bicarbonate).[5] Acute metabolic acidosis is associated with increased morbidity and mortality because of its depressive effects on cardiovascular function, increased risk of cardiac arrhythmias, stimulation of inflammation, and suppression of the immune response.[6] |
Evaluation of metabolic alkalosis | go to our full topic on Evaluation of metabolic alkalosis Metabolic alkalosis is an elevated arterial pH of above 7.45, and is the consequence of disorders that cause either a loss of hydrogen ions from the body or an increase in plasma bicarbonate from a normal value of 24 mEq/L. Causes include gastric secretion loss (e.g., vomiting) and mineralocorticoid excess. Patients may present with tingling, muscle cramps, weakness, cardiac arrhythmias, and/or seizures.[7][8] Some symptoms may be due to a decrease in circulating calcium, which occurs when the pH is high. Patients may develop serious or fatal arrhythmias and/or seizures without preceding symptoms. Compensatory metabolic alkalosis may be an incidental finding in patients with chronic respiratory acidosis. |
Evaluation of hyponatremia | go to our full topic on Evaluation of hyponatremia Defined as a serum sodium <135 mEq/L; severe hyponatremia is defined as a serum sodium <120 mEq/L. Hyponatremia is a common electrolyte disorder and is estimated to occur in 15% of all hospital inpatients.[9][10] Patients with hyponatremia have increased morbidity and mortality.[11] With few exceptions, when the serum sodium level is low, plasma osmolality is also low (hypotonic hyponatremia). While defined by the level of sodium, hypotonic hyponatremia is, in fact, a disorder of water balance. Common causes are administration of hypotonic fluids to patients and use of thiazide diuretics (more likely to affect older people).[12] Hyponatremia may also be a clue to the presence of serious underlying medical disorders. Patients who develop hyponatremia as a result of head injury, intracranial surgery, subarachnoid hemorrhage, stroke, or brain tumors may have cerebral salt-wasting syndrome or syndrome of inappropriate antidiuretic hormone (SIADH). A decrease in aldosterone production (e.g., Addison disease) causes an increase in sodium loss from the kidneys and hyponatremia. |
Evaluation of hypernatremia | go to our full topic on Evaluation of hypernatremia Hypernatremia is defined as a plasma sodium concentration of >145 mEq/L. Hypernatremia is a state of hyperosmolality, and is primarily a result of water deficit or sodium gain. Normally, persistently high sodium levels trigger antidiuretic hormone release, stimulating thirst mechanisms so that hypernatremia rarely develops. Hospitalized patients often have impaired thirst mechanisms, restricted access to water, and an increased risk of water loss (e.g., due to vomiting or fever). They are also at risk for iatrogenic inadequate fluid replacement. Endocrine abnormalities such as diabetes insipidus and mineralocorticoid excess may also lead to hypernatremia. Examination should focus on volume status. |
Evaluation of hypokalemia | go to our full topic on Evaluation of hypokalemia Hypokalemia is a serum potassium level <3.5 mEq/L. Clinical manifestations are typically seen only if the serum potassium level is <3.0 mEq/L, and include muscle weakness, ECG changes, cardiac arrhythmias, rhabdomyolysis, and renal abnormalities. Hypokalemia may result from decreased potassium intake, increased potassium entry into cells, increased potassium excretion (e.g., from the gastrointestinal tract, via urine or sweat), dialysis, or plasmapheresis. There are multiple causes of hypokalemia, including vomiting, severe diarrhea, laxative and bowel cleansing agent use in bulimia nervosa, chronic alcoholism, anorexia nervosa,[13] renal tubular acidosis,[14] primary aldosteronism, salt-wasting nephropathies,[15] and cystic fibrosis.[16] Some medications can cause hypokalemia, including diuretics, insulin treatment for diabetic ketoacidosis or nonketotic hyperglycemia, beta-adrenergic agonists such as albuterol or terbutaline, theophylline, chloroquine, laxative abuse or bowel-cleansing agent use, and administration of vitamin B12 or folic acid in megaloblastic anemia.[14] |
Evaluation of hyperkalemia | go to our full topic on Evaluation of hyperkalemia Significant hyperkalemia is defined as a serum potassium value >6.0 mEq/L. Moderate hyperkalemia is defined as serum potassium values in the 5.0 to 6.0 mEq/L range. Hyperkalemia can be life-threatening and may cause cardiac arrhythmias (ventricular fibrillation) by affecting the cardiac action potential. Hyperkalemia is often multifactorial in etiology. It may result from effective depletion of the circulating volume by heart failure combined with ACE inhibitors, or from increased dietary potassium intake combined with chronic renal failure. It is essential to take a thorough history of comorbidities and medications that might increase cellular potassium release or reduce urinary excretion. Reduced potassium excretion occurs in renal failure, volume depletion, and hypoaldosteronism.[17] Dietary factors (e.g., excess consumption of foods high in potassium) or medications may quickly lead to hyperkalemia in patients with comorbidities. |
Evaluation of hypocalcemia | go to our full topic on Evaluation of hypocalcemia Hypocalcemia is a state of electrolyte imbalance in which the circulating serum calcium level is low. Hypocalcemia arises mainly from either insufficient entry of calcium into the circulation or an increased loss of calcium from the circulation. There are multiple causes, including iatrogenic postsurgical hypoparathyroidism (usually transient), vitamin D deficiency, hypomagnesemia, hyperventilation, hypoparathyroidism, pseudohypoparathyroidism, hyperphosphatemia, hungry bone syndrome (rapid influx of calcium into the bones, causing more prolonged hypocalcemia following parathyroidectomy), acute pancreatitis, and can be drug-induced. It is also seen in critically ill patients. Hypocalcemia varies from a mild asymptomatic biochemical abnormality to a life-threatening disorder. Acute hypocalcemia can lead to paraesthesia, tetany, and seizures. Physical signs may be observed, including Chvostek sign (twitching of muscles innervated by the facial nerve). |
Evaluation of hypercalcemia | go to our full topic on Evaluation of hypercalcemia Symptoms from calcium elevation are typically not found unless the calcium is above 12 mg/dL. Severe hypercalcemia symptoms are more likely when calcium is >13 mg/dL. Hypercalcemia is harmful to the function of excitable membranes leading to skeletal muscle and gastrointestinal smooth muscle fatigue. Effects on cardiac muscle include a shortened QT interval and increased risk of cardiac arrest at very high calcium levels. Neurological sequelae include depression, irritability, and, with high enough levels, coma. High calcium may lead to precipitation in soft tissues such as the kidneys where renal function may be severely damaged. The most common causes of hypercalcemia are primary hyperparathyroidism and malignancy (e.g., multiple myeloma, leukemia, lung cancer, and breast cancer). Chronic symptoms are more consistent with hyperparathyroidism, whereas recent onset of symptoms suggests malignancy (the tumor is typically very advanced). Signs and symptoms include renal stones (typical of hyperparathyroidism), lethargy, easy fatigue, depression, irritability, constipation, gastrointestinal symptoms (e.g., nausea, vomiting, abdominal pain, peptic ulcer disease, pancreatitis), polyuria, polydipsia, confusion, and coma. Hypercalcemia may be asymptomatic.[18] |
Evaluation of magnesium deficiency | go to our full topic on Evaluation of magnesium deficiency Hypomagnesemia is defined as serum magnesium <1.8 mEq/L. Serum magnesium level is a poor indicator of the total magnesium content and availability in the body because only 1% of magnesium is found in the extracellular fluid. There is no simple, rapid, and accurate laboratory test to determine total body magnesium status in humans. Magnesium deficiency can be caused by decreased magnesium intake from the diet, decreased magnesium absorption, or increased renal magnesium excretion (renal magnesium wasting). Causes include malnutrition, isolated dietary magnesium deficiency, drug-induced, alcohol abuse, and pancreatitis. Symptoms are nonspecific and include: neuromuscular irritability similar to that produced by hypocalcemia, manifesting with extensor plantar reflexes, positive Trousseau and Chvostek signs, and, in severe cases, tetany; cardiovascular features such as rapid heartbeats and an elevated blood pressure, tachycardia, and/or ventricular arrhythmias; central nervous system symptoms of vertigo, ataxia, depression, and seizure activity. |
Primary hyperparathyroidism | go to our full topic on Primary hyperparathyroidism An endocrine disorder in which autonomous overproduction of parathyroid hormone (PTH) results in calcium metabolism derangement. Single parathyroid adenomas are the most common etiology (approximately 80% of cases) and familial forms are also well defined.[19] Multiple adenomas and hypertrophy of all 4 glands are less common. Diagnosis occurs through testing for a concurrent elevated serum calcium level and an inappropriately elevated intact serum PTH level. Inherited forms, affecting 10% to 20% of patients,[20] lead to hyperfunctioning parathyroid glands. Importantly, <1% of cases of hyperparathyroidism are caused by parathyroid carcinoma. In 2017, normocalcemic primary hyperparathyroidism was recognized. It presents with high levels of PTH but with normal serum and ionized calcium levels. Some, but not all, patients will go on to develop primary hyperparathyroidism.[21] Complications due to primary PTH are uncommon and include osteoporosis and bone fracture due to leaching of calcium from bones, and renal calculi due to elevated serum and urine calcium. |
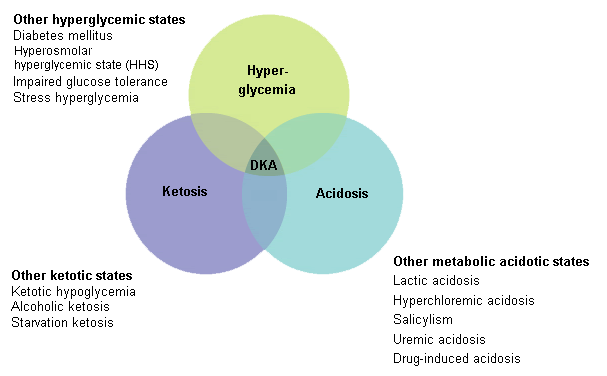
Diabetic ketoacidosis | go to our full topic on Diabetic ketoacidosis An acute metabolic complication of diabetes mellitus that is potentially fatal and requires prompt medical attention for successful treatment. Diabetic ketoacidosis (DKA) is characterized by a biochemical triad of hyperglycemia, ketonemia, and metabolic acidosis, with rapid symptom onset. DKA may be the first presentation of diabetes. In DKA, there is a reduction in the net effective concentration of circulating insulin along with an elevation of counter-regulatory hormones (glucagon, catecholamines, cortisol, and growth hormone). DKA is usually characterized by plasma glucose >250 mg/dL, arterial pH 7.0 to <7.3, and the presence of ketonemia and/or ketonuria. Serum sodium, chloride, magnesium, and calcium are usually low; serum anion gap is elevated; and serum potassium, urea, and creatinine are usually elevated. Arterial bicarbonate ranges from <5 mEq/L in severe DKA to <15 mEq/L in mild DKA. Venous pH is recommended for monitoring treatment. Rarely, patients present with euglycemic DKA and have a normal blood glucose level. [Figure caption and citation for the preceding image starts]: Triad of DKAAdapted with permission from: Kitabchi AE, Wall BM. Diabetic ketoacidosis. Med Clin North Am. 1995;79:9-37 [Citation ends]. |
Hyperosmolar hyperglycemic state | go to our full topic on Hyperosmolar hyperglycemic state A serious metabolic complication of diabetes mellitus characterized by profound hyperglycemia (glucose >600 mg/dL), hyperosmolality (effective serum osmolality >320 mOsm/kg), and volume depletion in the absence of significant ketoacidosis (pH >7.3 and hydrogen carbonate [bicarbonate] >15 mEq/L).[22] It is most common in older patients with type 2 diabetes mellitus. Contributes to less than 1% of all diabetes-related admissions. However, mortality is high: at approximately 15%.[23] Infection is the major precipitating factor, occurring in 30% to 60% of patients. Urinary tract infections and pneumonia are the most common infections reported.[22][24] Acute cognitive impairment (lethargy, disorientation, stupor) is common and correlates best with effective serum osmolality. Coma is rare and, if seen, is usually associated with a serum osmolality >340 mOsm/kg (>340 mmol/kg).[23] |
Renal tubular acidosis | go to our full topic on Renal tubular acidosis The term renal tubular acidosis (RTA) refers to a group of renal disorders in which there are defects in the reabsorption of bicarbonate or the excretion of hydrogen ions, or both. The acid retention or bicarbonate loss results in the development of hyperchloremic metabolic acidosis. Therefore the RTA syndromes are characterized by a relatively normal glomerular filtration rate and metabolic acidosis accompanied by hyperchloremia and a normal anion gap.[25] Adult patients with RTA are often asymptomatic but may present with muscular weakness related to associated hypokalemia, nephrocalcinosis, or recurrent renal stones. Proximal and classic distal RTA are characterized by hypokalemia.[25][26] Hyperkalemia in distal RTA indicates that aldosterone deficiency or resistance is the cause of the problem.[25] There is minimal or absent urine ammonium in hyperkalemic distal RTA. Serum sodium is usually normal. RTA is rarely symptomatic. Patients with severe acidemia can show hyperventilation or Kussmaul breathing due to respiratory compensation. The urine pH exceeds 5.5 in classic distal RTA, but is lower than 5.0 in patients with untreated proximal RTA. |
Primary aldosteronism | go to our full topic on Primary aldosteronism Aldosterone’s primary function is to regulate sodium absorption and potassium excretion in the distal tubule. In primary aldosteronism (PA), aldosterone production exceeds the body's requirements and is relatively autonomous with regard to its normal chronic regulator, the renin-angiotensin II (AII) system.[27][28] This results in excessive sodium reabsorption via the distal nephron, leading to an increase in the volume of water taken up through the nephron contributing to the development of hypertension and suppression of renin-AII. Urinary loss of potassium and hydrogen ions, exchanged for sodium at the distal nephron, may result in hypokalemia and metabolic alkalosis if severe and prolonged, however, most of patients are normokalemic.[27][28] Primary aldosteronism is the most common specifically treatable and potentially curable form of hypertension, accounting for at least 5% of hypertensive patients. Approximately 30% of people with PA have unilateral forms, and 70% have bilateral forms.[29] |
Primary adrenal insufficiency (Addison disease) | go to our full topic on Primary adrenal insufficiency (Addison disease) A disorder that results from intrinsic diseases that affect the cortex of the adrenal glands, causing impairment in the synthesis and secretion of all steroids (glucocorticoids and/or mineralocorticoids) normally secreted by the three cortical layers.[30] Approximately 90% of the adrenal cortex needs to be destroyed to result in adrenal insufficiency. Primary adrenal insufficiency (PAI) is a rare condition with an overall prevalence of about 100 to 140 cases per million and an incidence of 4:1,000,000 per year in developed countries.[31] It may be acute (adrenal crisis) or insidious. PAI presents with substantial fatigue and generalized weakness associated with mucocutaneous hyperpigmentation, hypotension and/or postural hypotension, and salt craving. The finding of low sodium and high potassium serum levels is typical. If untreated, it is a potentially life-threatening condition. Adrenocorticotropic hormone stimulation test is performed to confirm or exclude the diagnosis of PAI. |
Syndrome of inappropriate antidiuretic hormone | go to our full topic on Syndrome of inappropriate antidiuretic hormone Syndrome of inappropriate antidiuretic hormone (SIADH) is defined as euvolemic, hypotonic hyponatremia secondary to impaired free water excretion, usually from excessive antidiuretic hormone (ADH) secretion either from the pituitary or more commonly a nonpituitary source which may include medication or cancer (lung malignancy is the most common). ADH, also known as arginine vasopressin, facilitates free water absorption in the collecting tubule. Under pathologic conditions, the pituitary gland and other cells may synthesize and secrete ADH independently of serum osmolality or circulating volume.[9] SIADH is primarily identified by abnormal serum sodium levels on laboratory testing, but patients may present with signs of cerebral edema, including nausea, vomiting, headache, mental status changes, increased somnolence, or coma, and appear euvolemic. |
Contributors
Authors
Editorial Team
BMJ Publishing Group
Disclosures
This overview has been compiled using the information in existing sub-topics.
References
Key articles
Berend K, de Vries AP, Gans RO. Physiological approach to assessment of acid-base disturbances. N Engl J Med. 2014 Oct 9;371(15):1434-45. Abstract
Seifter JL. Integration of acid-base and electrolyte disorders. N Engl J Med. 2014 Nov 6;371(19):1821-31. Abstract
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Use of this content is subject to our disclaimer