Resumo
Definição
História e exame físico
Principais fatores diagnósticos
- história familiar de polipose adenomatosa familiar (PAF)/PAF atenuada
- adolescentes
- início de câncer colorretal na meia idade
- pigmentação bilateral da retina
Outros fatores diagnósticos
- constipação/diarreia
- hematoquezia
- características extraintestinais da polipose adenomatosa familiar (PAF)
Fatores de risco
- mutação das linhas germinativas no gene da polipose adenomatosa do cólon (APC)
- história familiar de polipose adenomatosa familiar (PAF) ou PAF atenuada
Investigações diagnósticas
Primeiras investigações a serem solicitadas
- teste genético
Algoritmo de tratamento
PAF: sem adenomas colônicos
PAF: com adenomas colônicos
PAF atenuada: sem adenomas colônicos
PAF atenuada: com adenomas colônicos
Colaboradores
Autores
Priyanka Kanth, MD, MS, FACG, AGAF
Associate Professor of Medicine
Division of Gastroenterology
MedStar Georgetown University Hospital
Lombardi Comprehensive Cancer Center
Washington
DC
Declarações
PK declares that she has no competing interests.
Agradecimentos
Dr Priyanka Kanth would like to gratefully acknowledge Dr Charles A. Ternent, Dr Alan G. Thorson, Dr Lisa A. Boardman, and Dr Douglas L. Riegert-Johnson, the previous contributors to this topic.
Declarações
CAT, AGT, LAB, and DLRJ declare that they have no competing interests.
Revisores
Galen Leung, MD
Assistant Professor of Clinical Medicine
Perelman School of Medicine
University of Pennsylvania
Philadelphia
PA
Declarações
GL declares that he has no competing interests.
Jatin Roper, MD
Assistant Professor of Medicine
Duke University School of Medicine
Duke University
Durham
NC
Declarações
JR declares that he is a consultant for Microbial Machines.
Yann Parc, MD, PhD
Professor of General Surgery
Department of Digestive Surgery
Hopital Saint-Antoine
Universite Pierre et Marie Curie Paris VI
Paris
France
Declarações
YP declares that he has no competing interests.
Gabriela Moslein, MD
Editorial Board
Allgemein- und Viszeralchirurgie
St Josefs-Hospital Bochum-Linden
Dusseldorf
Germany
Declarações
GM declares that she has no competing interests.
Créditos aos pareceristas
Os tópicos do BMJ Best Practice são constantemente atualizados, seguindo os desenvolvimentos das evidências e das diretrizes. Os pareceristas aqui listados revisaram o conteúdo pelo menos uma vez durante a história do tópico.
Declarações
As afiliações e declarações dos pareceristas referem--se ao momento da revisão.
Referências
Principais artigos
National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: genetic/familial high-risk assessment: colorectal, endometrial, and gastric [internet publication].Texto completo
Yang J, Gurudu SR, Koptiuch C, et al. American Society for Gastrointestinal Endoscopy guideline on the role of endoscopy in familial adenomatous polyposis syndromes. Gastrointest Endosc. 2020 May;91(5):963-82.e2.Texto completo Resumo
Hyer W, Cohen S, Attard T, et al. Management of familial adenomatous polyposis in children and adolescents: Position paper from the ESPGHAN polyposis working group. J Pediatr Gastroenterol Nutr. 2019 Mar;68(3):428-41.Texto completo Resumo
Poylin VY, Shaffer VO, Felder SI, et al. The American Society of Colon and Rectal Surgeons Clinical Practice Guidelines for the management of inherited adenomatous polyposis syndromes. Dis Colon Rectum. 2024 Feb 1;67(2):213-27.Texto completo Resumo
Zaffaroni G, Mannucci A, Koskenvuo L, et al. Updated European guidelines for clinical management of familial adenomatous polyposis (FAP), MUTYH-associated polyposis (MAP), gastric adenocarcinoma, proximal polyposis of the stomach (GAPPS) and other rare adenomatous polyposis syndromes: a joint EHTG-ESCP revision. Br J Surg. 2024 May 3;111(5):znae070.Texto completo Resumo
Artigos de referência
Uma lista completa das fontes referenciadas neste tópico está disponível para os usuários com acesso total ao BMJ Best Practice.
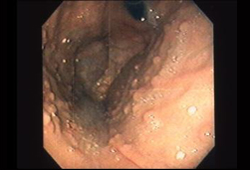
Diagnósticos diferenciais
- Polipose MutYH
- Polipose juvenil
- síndrome de Peutz-Jeghers
Mais Diagnósticos diferenciaisDiretrizes
- Suspected cancer: recognition and referral
- Clinical practice update on nonampullary duodenal lesions: expert review
Mais DiretrizesFolhetos informativos para os pacientes
Câncer de intestino: o que é?
Câncer de intestino: devo fazer rastreamento?
Mais Folhetos informativos para os pacientes Conectar-se ou assinar para acessar todo o BMJ Best Practice
Conectar-se ou assinar para acessar todo o BMJ Best Practice
O uso deste conteúdo está sujeito ao nosso aviso legal