Resumen
Definición
Anamnesis y examen
Principales factores de diagnóstico
- presencia de factores de riesgo
- Antecedentes familiares
- inicio en la infancia (enfermedades de mucopolisacaridosis [MPS], Pompe, Gaucher, Fabry, Niemann-Pick tipo A)
- inicio en la adolescencia (Fabry, Pompe, Gaucher tipos 1, 3, mucopolisacaridosis, Niemann-Pick tipos B, C)
- inicio en la edad adulta (enfermedades de Fabry, Gaucher tipo 1, Pompe)
- hepatomegalia y/o esplenomegalia
- hiperacusia
- antecedentes de insuficiencia renal
- erupción cutánea/lesiones cutáneas
- circunferencia de la cabeza grande
- mancha macular de color "rojo cereza" en la oftalmoscopia
- atrofia óptica o retinitis pigmentaria en la oftalmoscopia
- opacidad corneal en la oftalmoscopia
- fatiga
Otros factores de diagnóstico
- retraso en el desarrollo neurológico
- deterioro auditivo/sordera repentina
- catarata en la oftalmoscopia
- trastorno del movimiento ocular
- demencia progresiva y ataxia o alteración de la marcha
- retraso en el desarrollo
- contractura articular
- depresión
- alteraciones óseas, incluido giba de la columna
- hidrocefalia
- antecedentes de infecciones recurrente de las vías respiratorias
- psicosis
- trastornos del movimiento
- accidente cerebrovascular/ataque isquémico transitorio precoz
- cardiomegalia
- valvulopatía cardíaca
Factores de riesgo
- sexo masculino (mucopolisacaridosis [MPS] II, enfermedad de Fabry)
- etnia asquenazí
Pruebas diagnósticas
Primeras pruebas diagnósticas para solicitar
- análisis enzimático
- análisis del sustrato
- análisis de ADN
- hemograma completo (HC)
Pruebas diagnósticas que deben considerarse
- electrocardiograma (ECG)
- ecocardiograma
- pruebas funcionales respiratorias
- biopsia de médula ósea
- biopsia muscular
- tomografía computarizada (TC)/IRM (resonancia magnética) de órgano agrandado (enfermedad de Gaucher)
- ecografía/IRM (Fabry)
- tomografía computarizada (TC)/radiografía (mucopolisacaridosis)
Algoritmo de tratamiento
enfermedad de Gaucher tipo 1
enfermedad de Gaucher tipo 2
enfermedad de Gaucher tipo 3
Enfermedad de Fabry
mucopolisacaridosis (MPS)
Enfermedad de Pompe
enfermedad de Tay-Sachs
enfermedad de Niemann-Pick
Colaboradores
Autores
Atul B. Mehta, MA, MB BChir, MD, FRCP, FRCPath
Consultant Haematologist
Emeritus Professor in Haematology
University College London
London
UK
Divulgaciones
ABM has participated in educational activities for Sanofi Genzyme, Takeda and Amicus including advisory boards for which he has received honoraria and travel grants. He has also received research funding from Sanofi Genzyme, Takeda and Amicus. He is also an author of a number of references cited in this topic.
Revisores por pares
Gregory M. Pastores, MD
Associate Professor
Departments of Neurology and Pediatrics
NYU School of Medicine
New York, NY
Divulgaciones
GMP declares that he has no competing interests.
Uma Ramaswami, MD, FRCPCH
Consultant Paediatrician
Paediatric Metabolic Unit
Cambridge University Hospitals
Cambridge
UK
Divulgaciones
UR has received travel grants, honoraria for lectures, and funding for clinical trials from Shire HGT, Genzyme, and Actelion.
Elmer V. Villanueva, MD, ScM, FRIPH
Associate Professor of Public Health
Director of Research
Gippsland Medical School
Monash University
Churchill
Australia
Divulgaciones
EVV declares that he has no competing interests.
Agradecimiento de los revisores por pares
Los temas de BMJ Best Practice se actualizan de forma continua de acuerdo con los desarrollos en la evidencia y en las guías. Los revisores por pares listados aquí han revisado el contenido al menos una vez durante la historia del tema.
Divulgaciones
Las afiliaciones y divulgaciones de los revisores por pares se refieren al momento de la revisión.
Referencias
Artículos principales
Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 2017 Feb 17;18(2):441.Texto completo Resumen
Germain DP. Fabry disease. Orphanet J Rare Dis. 2010 Nov 22;5:30.Texto completo Resumen
Patterson MC, Clayton P, Gissen P, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol Clin Pract. 2017 Dec;7(6):499-511.Texto completo Resumen
Artículos de referencia
Una lista completa de las fuentes a las que se hace referencia en este tema está disponible para los usuarios con acceso a todo BMJ Best Practice.
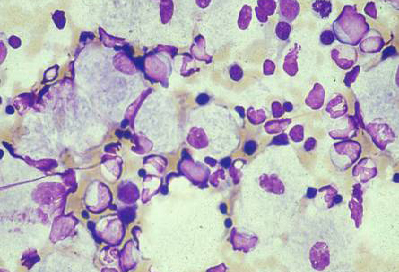
Diferenciales
- Histiocitosis de células de Langerhans (diagnóstico diferencial de la enfermedad de Gaucher de tipo 2 y 3)
- Fiebre reumática (diagnóstico diferencial de la enfermedad de Fabry)
- Endocarditis bacteriana (diagnóstico diferencial de la enfermedad de Fabry)
Más DiferencialesGuías de práctica clínica
- Pegunigalsidase alfa for treating Fabry disease
- Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann-Pick disease types A, B and A/B)
Más Guías de práctica clínica Inicie sesión o suscríbase para acceder a todo el BMJ Best Practice
Inicie sesión o suscríbase para acceder a todo el BMJ Best Practice
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad