Summary
Definition
DiGeorge syndrome, also known as 22q11.2 deletion syndrome (22q11.2DS) or velocardiofacial syndrome, is a condition that describes characteristic, but variable, morphologic and neurologic features that result from the deletion of one copy of 22q11.2.[1] The classic presentation is a triad of cardiac anomalies, hypoplastic thymus, and hypocalcemia (resulting from parathyroid hypoplasia). The deletion causes a reduction in TBX1, a key transcription factor for development of the pharyngeal arches.[2] This developmental disruption may cause cardiac anomalies, immunologic abnormalities, cleft lip and palate, hypoparathyroidism, learning disabilities, and schizophrenia.[3] The disorder is notable for marked variation in the penetrance of the various features. The syndrome has been known by multiple other names, including CATCH22 and Shprintzen syndromes. The complexity of the nomenclature is due to great variability in the clinical syndrome.
The phenotype of DiGeorge syndrome may be divided into two components. The first, pharyngeal component consists of congenital heart disease, hypoplasia of the parathyroid glands, thymic hypoplasia with T-cell immunodeficiency, cleft lip and palate, and mild dysmorphic facial features. The second, neurologic phenotype consists of mild cognitive dysfunction, which typically presents as learning disabilities, speech impairment, and an increased incidence of schizophrenia.[Figure caption and citation for the preceding image starts]: Loss of 1 copy of 22q11.2, demonstrated by microarray copy number analysisFrom the collections of Sean A. McGhee, MD and Maria Garcia Lloret, MD [Citation ends].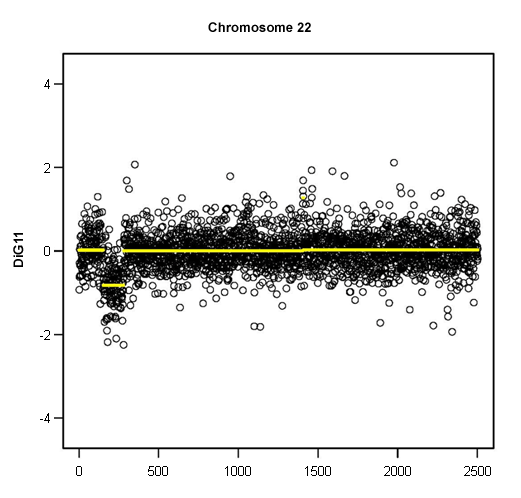
History and exam
Key diagnostic factors
- cyanosis
- signs of heart failure
- heart murmur
- characteristic facial features
- cleft lip and palate
- faltering growth
- seizure or tetany
Other diagnostic factors
- presentation in infancy
- feeding difficulty
- speech delay
- nonverbal learning disorder
- frequent infections
- schizophrenia
- features of CHARGE syndrome
Risk factors
- parent with DiGeorge syndrome
Diagnostic tests
1st tests to order
- serum calcium
- serum intact PTH
- T-cell count
- chromosomal microarray analysis
- multiplex ligation-dependent probe amplification
- immune-specific titers (if previously immunized)
- CBC
- TSH
- free T4
- chest x-ray
- ECG
- serum immunoglobulins
Tests to consider
- fluorescence in situ hybridization
- karyotype
- echocardiogram
- renal and bladder ultrasound
- audiometry
- dental and palatal evaluations
- ophthalmology evaluation
- spinal x-rays
Treatment algorithm
birth to 4 months
infants and toddlers (4 months to 5 years)
school-aged children (5 to 18 years)
adults
Contributors
Authors
James A. Stadler III, MD, MAS
Assistant Professor of Neurological Surgery
University of Wisconsin School of Medicine and Public Health
University of Wisconsin-Madison
WI
Disclosures
JAS declares that he has no competing interests.
Acknowledgements
Dr James A. Stadler III would like to gratefully acknowledge Dr Sean A. McGhee and Dr Maria Garcia Lloret, the previous contributors to this topic.
Disclosures
SAMG has provided testimony as an expert in a criminal case in which immune deficiency was a concern, and is a co-author of a reference cited in this topic. MGL is a co-author of a reference cited in this topic.
Peer reviewers
Gabriela M. Repetto, MD
Director
Centro de Genetica Humana
Facultad de Medicina
Clínica Alemana-Universidad del Desarrollo
Santiago
Chile
Disclosures
GMR is a co-author of a reference cited in this topic.
Lisa Kobrynski, MD, MPH
Associate Professor of Pediatrics
Division of Pulmonary, Allergy & Immunology, Cystic Fibrosis, and Sleep
Emory University
Atlanta
GA
Disclosures
LK is an investigator in clinical trials by Baxter Bioscience. These trials do not involve patients with 22q11DS. LK is an author of a number of references cited in this topic.
Winnie Ip, BHB, MBChB, FRACP, MD(Res)
Consultant Immunologist
Department of Immunology
Great Ormond Street Hospital
London
UK
Disclosures
WI declares that she has no competing interests.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
European Society for Immunodeficiencies. Clinical Working Party diagnostic criteria for PID: DiGeorge syndrome diagnostic criteria [internet publication].Full text
Boot E, Óskarsdóttir S, Loo JCY, et al. Updated clinical practice recommendations for managing adults with 22q11.2 deletion syndrome. Genet Med. 2023 Mar;25(3):100344.Full text Abstract
Óskarsdóttir S, Boot E, Crowley TB, et al. Updated clinical practice recommendations for managing children with 22q11.2 deletion syndrome. Genet Med. 2023 Mar;25(3):100338.Full text Abstract
Mustillo PJ, Sullivan KE, Chinn IK, et al. Clinical practice guidelines for the immunological management of chromosome 22q11.2 deletion syndrome and other defects in thymic development. J Clin Immunol. 2023 Feb;43(2):247-70.Full text Abstract
Scheuerle AE, Geleske TA, Merchant N, et al. Health supervision for children with 22q11.2 deletion syndrome: clinical report. Pediatrics. 2025 Aug 1;156(2):e2025072717.Full text Abstract
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Differentials
- Nonsyndromic anomalies
- Isotretinoin exposure
- CHARGE syndrome
More DifferentialsGuidelines
- Updated clinical practice recommendations for managing children with 22q11.2 deletion syndrome
- Clinical practice guidelines for the immunological management of chromosome 22q11.2 deletion syndrome and other defects in thymic development
More GuidelinesPatient information
Heart failure
Schizophrenia: what is it?
More Patient information Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer