Summary
Definition
History and exam
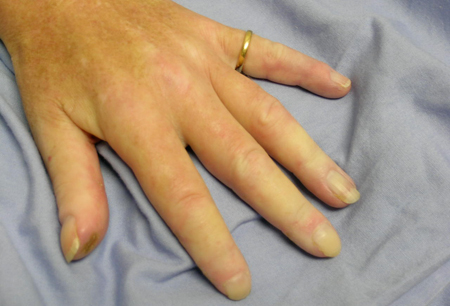
Key diagnostic factors
- Raynaud phenomenon (RP)
- dysphagia
- GERD
- dilated nailbed capillaries
- bright shiny skin of hands, feet
- symmetrical swelling, tight fingers
- prayer sign
- claw hand deformities
- ulcers on fingers
- flexion contracture of hands
- carpal tunnel syndrome
- calcinosis
- large well-demarcated telangiectasia
Other diagnostic factors
- skin pigmentation changes
- family history of connective tissue disease
Risk factors
- silica dust exposure
- family history of scleroderma
- polyvinyl chlorides
Diagnostic tests
1st tests to order
- complete blood count
- serum creatinine
- serum antinuclear antibody
- serum extractable nuclear antigens
- serum erythrocyte sedimentation rate (ESR)
- echocardiogram
- pulmonary function tests (PFTs)
Tests to consider
- peripheral blood smear
- anticentromere antibody
- high-resolution CT chest
- esophageal manometry
Treatment algorithm
renal crisis
no renal crisis
Contributors
Authors
Janet E. Pope, MD, MPH, FRCPC

Professor of Medicine
Division of Rheumatology
Department of Medicine
Schulich School of Medicine and Dentistry
University of Western Ontario
Head
Division of Rheumatology
St. Joseph's Health Care
London
Ontario
Canada
Disclosures
JEP has consulted for and/or performed research trials with AbbVie, Amgen, AstraZeneca, BI, BMS, Fresenius Kabi, Galapagos, GSK, Janssen, Lilly, Merck, Novartis, Organon, Pfizer, Roche, Sandoz, Sanofi, United Chemicals Belgium, Horizon, Mallinckrodt, and Viatris. JEP is an author of several references cited in this topic.
Peer reviewers
Virginia Steen, MD
Professor of Medicine
Georgetown University
Washington
DC
Disclosures
VS has received research funding, consulting fees, and honorarium from Actelion and Gilead, which could result in competing interests. VS is an author of several references cited in this topic.
Frank Wollheim, MD
Emeritus Professor
Department of Rheumatology
Lund University Hospital
Lund
Sweden
Disclosures
FW is an author of a reference cited in this topic.
Gabriela Riemekasten, PD, Dr. med.
Charité-Universitätsmedizin Berlin
Medizinische Klinik mit Schwerpunkt
Rheumatologie und Klinische Immunologie
Charitéplatz
Berlin
Germany
Disclosures
GR is a consultant for Encysive, Actelion, Bayer, GSK, Roche, and other companies, and received lecture fees from all of these companies involved in systemic scleroderma. GR is an author of a reference cited in this topic.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
Mayes MD, Lacey JV Jr., Beebe-Dimmer J, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003 Aug;48(8):2246-55.Full text Abstract
van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis. 2013 Nov;72(11):1747-55.Full text Abstract
Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017 Aug;76(8):1327-39.Full text Abstract
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Differentials
- Generalized morphea
- Eosinophilic fasciitis (Shulman syndrome)
- Nephrogenic systemic fibrosis
More DifferentialsGuidelines
- Treatment of systemic sclerosis-associated interstital lung disease: evidence-based recommendations
- 2022 Guideline for vaccinations in patients with rheumatic and musculoskeletal diseases
More GuidelinesPatient information
Carpal tunnel syndrome
Raynaud phenomenon
More Patient information Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer