Summary
Definition
History and exam
Key diagnostic factors
- family history of Marfan disease
- tall stature
- wide arm span
- high level of pubic bone
- high arched palate
- arachnodactyly
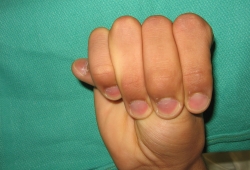
- positive wrist sign
- positive thumb sign
- pectus excavatum (funnel chest)
- pectus carinatum (pigeon breast)
- scoliosis
- flat feet (pes planus)
- dislocated/subluxed eye lens
- myopia and/or astigmatism
- retinal abnormalities
- joint hypermobility
- aortic valve murmur
- mitral valve murmur
- history of treatment for dental crowding
- history of myopia and/or astigmatism
- reduced elbow extension
Other diagnostic factors
- glaucoma
- history of spontaneous pneumothorax
- striae
- low back ache
- joint pain
- inguinal/abdominal/incisional hernias
- dyspnea
- signs of heart failure
Risk factors
- family history of Marfan syndrome
- family history of aortic dissection or aneurysm
- high paternal age
Diagnostic tests
1st tests to order
- echocardiography
- slit-lamp eye exam with intraocular pressure measurement
- CT scan, thorax
- MRI, thorax
Tests to consider
- blood screening for fibrillin-1 (FBN-1) gene mutation
- CXR
- CT scan, abdomen
- MRI, abdomen
- ultrasound, abdomen
- CT scan, lower spine
- MRI, lower spine
- plasma homocysteine
Treatment algorithm
aortic dilation meeting the indications for surgery
retinal tear
retinal detachment
aortic dilation not meeting indications for surgery or following aortic surgery
myopia
lens subluxation/dislocation
cataract
scoliosis/kyphoscoliosis
severe pectus excavatum/carinatum with evidence of cardiopulmonary compromise
arthropathy and/or spondylolisthesis
Contributors
Authors
Anne Child, MD, FRCP
Medical Director Marfan Trust
Honorary Consultant Royal Brompton Group GSTT NHS Foundation Trust
Keston
Kent
UK
Disclosures
AC is an author of several references cited in this topic but has no competing interests.
Acknowledgements
Dr Anne Child would like to gratefully acknowledge Dr Aman Chandra and Dr Maite Tome, the previous contributors to this topic. Dr Anne Child and Dr Aman Chandra would also like to thank Anna Apara for her work on this topic.
Disclosures
AC, MT, and AA declare that they have no competing interests.
Peer reviewers
Daniel Judge, MD
Assistant Professor of Medicine
Medical Director
JHU Center for Inherited Heart Disease
Johns Hopkins Hospital
Baltimore
MD
Disclosures
DJ declares that he has no competing interests.
Reed E. Pyeritz, MD, PhD
Professor of Medicine and Genetics
University of Pennsylvania
Philadelphia
PA
Disclosures
REP declares that he has no competing interests.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
Tinkle BT, Lacro RV, Burke LW, et al. Health supervision for children and adolescents with Marfan syndrome. Pediatrics. 2023 Apr 1;151(4):e2023061450. Abstract
Isselbacher EM, Preventza O, Black JH 3rd, et al. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease: a report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2022 Dec 13;146(24):e334-482.Full text Abstract
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Differentials
- Aortic dissection not associated with Marfan syndrome
- Bicuspid aortic valve
- Ehlers-Danlos syndrome
More DifferentialsGuidelines
- Guideline for the diagnosis and management of aortic disease
- 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases
More Guidelines Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer