Summary
Definition
History and exam
Key diagnostic factors
- cognitive impairment
- limb and/or gait ataxia
- myoclonus
- parkinsonism
- psychiatric symptoms
- visual changes
- age late 20s or mid-to-late 60s
- insomnia, dysautonomia
- positive family history
- nonspecific or constitutional symptoms
Other diagnostic factors
- painful sensory symptoms
- movement disorder
Risk factors
- genetic predisposition
- prion-contaminated surgical instruments
- transfusion of blood or blood products (variant Creutzfeldt-Jakob disease)
- consumption of UK beef from 1980 to 1996
- consumption of US beef
- deer, elk, moose hunting in endemic regions of US and Canada
- use of human growth hormone
Diagnostic tests
1st tests to order
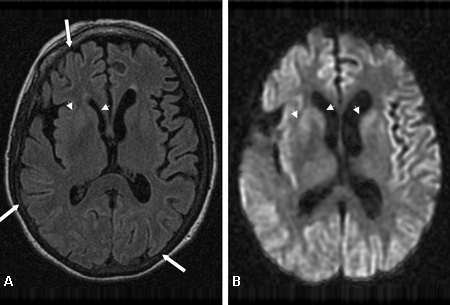
- brain MRI
- EEG
Tests to consider
- quaking-induced conversion (QuIC)
- CSF biomarkers
- prion protein gene genetic testing
- biopsy (brain, tonsil)
Treatment algorithm
all patients
Contributors
Authors
Valerie Sim, MD, FRCPC
Associate Professor
Department of Medicine
Division of Neurology
Centre for Prions and Protein Folding Diseases
University of Alberta
Edmonton
Alberta
Canada
Disclosures
VS is principal and co-investigator on several research grants through the University of Alberta, to study prion disease and related neurodegenerative diseases. VS has one patent approved for the development of IgE-based prion therapy. VS is currently a site lead for the Ionis PrProfile trial. VS has been invited to give an educational session on prion disease at the American Academy of Neurology meeting in 2024.
Acknowledgements
Dr Valerie Sim would like to gratefully acknowledge Dr Michael D. Geschwind, Dr Amy Kuo, and Dr R. Ronald Finley, the previous contributors to this topic. MDG participates in the speakers' bureau for Pfizer, Forest, and Novartis; is consultant for MedaCorp, Gerson-Lehman Group, and Clinical Advisors Incorporated; and is an author of a number of references cited in this topic. RRF participates in the speakers' bureau for Pfizer, Forest, and Novartis. AK declares that she has no competing interests.
Peer reviewers
Ali Hassoun, MD, FACP, FIDSA, AAHIVS
Infectious Disease Specialist
Alabama Infectious Diseases Center
Huntsville
AL
Disclosures
AH declares that he has no competing interests.
Robert A. Larsen, MD
Associate Professor of Medicine
University of Southern California
Keck School of Medicine
Los Angeles
CA
Disclosures
RAL declares that he has no competing interests.
William A. Petri, Jr., MD, PhD, FACP
Chief and Professor of Medicine
Division of Infectious Diseases and International Health
University of Virginia Health System
Charlottesville
VA
Disclosures
WAP declares that he has no competing interests.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
Department of Health. Minimise transmission risk of CJD and vCJD in healthcare settings. Nov 2021 [internet publication].Full text
Centers for Disease Control and Prevention. BSE (Bovine spongiform encephalopathy, or mad cow disease). Sep 2021 [internet publication].Full text
American College of Radiology. ACR appropriateness criteria: movement disorders and neurodegenerative diseases. 2019 [internet publication].Full text
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Differentials
- Alzheimer dementia (AD)
- Lewy body dementia
- Frontotemporal dementia
More DifferentialsGuidelines
- ACR appropriateness criteria: movement disorders and neurodegenerative diseases
- Evidence-based guideline: diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease
More Guidelines Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer