Summary
Definition
History and exam
Key diagnostic factors
- family history of NF1
- pain, any location
- neurologic deficits: gross motor delay, general incoordination, school performance problems
- compromised vision
- compromised social interactions
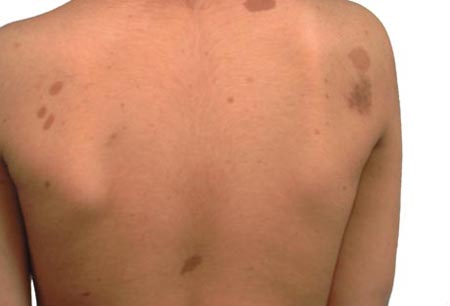
- skin: café au lait spots, axillary freckling, cutaneous juvenile xanthogranulomas, neurofibromas
- head and neck: unilateral diffuse plexiform neurofibroma divisions of the trigeminal nerve
- ophthalmologic: visual compromise, optic disk pallor, iris Lisch nodules, choroidal abnormalities
- central nervous system: signs of hydrocephalus, brain tumors, and/or cerebellar abnormalities
- peripheral nervous system: palpable mass about neck, brachial plexuses, groin, Hunter canal, or the popliteal fossae
- skeletal: tibial dysplasia or pseudarthrosis, sphenoid wing dysplasia, pectus excavatum or carinatum, genu valgum or varum, ankle valgus, pes planus
- gastrointestinal: severe constipation, obstipation, abdominal pain, gastrointestinal bleeding
- vascular: neurologic problems, abdominal pain (and/or hemorrhage)
- autism spectrum disorder
- vascular: hypertension
Risk factors
- parent with type 1 neurofibromatosis (NF1)
Diagnostic tests
1st tests to order
- clinical diagnosis
Tests to consider
- genetic testing to confirm type 1 neurofibromatosis mutation
- MRI and/or CT scans
- PET scan
- biopsy
Treatment algorithm
pheochromocytoma
malignant peripheral nerve sheath tumor or atypical neurofibromatous neoplasm of uncertain biologic potential
neurofibromas: noncutaneous
cutaneous
headache
nervous system
eye
oral
skeletal
vascular
gastrointestinal
hematopoietic
psychological
pregnancy-related
Contributors
Authors
D. Gareth Evans, MD, FRCP
Professor of Medical Genetics and Cancer Epidemiology
Genomic Medicine
School of Medicine
University of Manchester
Manchester
UK
Disclosures
DGE declares he has no conflicts of interest.
Acknowledgements
Professor D. Gareth Evans would like to gratefully acknowledge Dr Vincent M. Riccardi, a previous contributor to this topic, and Dr Grace R. Vassallo and Dr Helen Young who provided expert input on the management of type 1 neurofibromatosis.
Disclosures
VMR and HY declare that they have no competing interests. GRV has been reimbursed for providing advisory work to Alexion, sponsored by Alexion to attend conferences, and reimbursed for providing teaching for Alexion. She has also provided advisory work to Merck, and has provided paid interviews for Atheneum.
Peer reviewers
Laura Klesse, MD, PhD
Professor of Pediatrics and Neurological Surgery
UT Southwestern Medical Center Dallas
Dallas
TX
Disclosures
LK has taken part in medical advisory boards for SpringWorks and Alexion.
Patrick Morrison, MD
Consultant in Clinical Genetics
Department of Medical Genetics
Belfast HSC Trust
Belfast
UK
Disclosures
PM declares that he has no competing interests.
Edward S. Tobias, BSc (Hons), MBChB, MRCP (UK), PhD
Clinical Senior Lecturer and Honorary Consultant in Medical Genetics
Institute of Medical Genetics
Yorkhill Hospital
University of Glasgow
Scotland
UK
Disclosures
EST declares that he has no competing interests.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
Miller DT, Freedenberg D, Schorry E, et al; Council on Genetics; American College of Medical Genetics and Genomics. Health supervision for children with neurofibromatosis type 1. Pediatrics. 2019 May;143(5):e20190660.Full text Abstract
Legius E, Messiaen L, Wolkenstein P, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. 2021 Aug;23(8):1506-13.Full text Abstract
Carton C, Evans DG, Blanco I, et al. ERN GENTURIS tumour surveillance guidelines for individuals with neurofibromatosis type 1. EClinicalMedicine. 2023 Jan 13;56:101818.Full text Abstract
Perrino MR, Das A, Scollon SR, et al. Update on pediatric cancer surveillance recommendations for patients with neurofibromatosis type 1, Noonan syndrome, CBL syndrome, Costello syndrome, and related RASopathies. Clin Cancer Res. 2024 Nov 1;30(21):4834-43.Full text Abstract
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Differentials
- NF2-related schwannomatosis (NF2)
- McCune-Albright syndrome
- Familial café au lait spots
More DifferentialsGuidelines
- Consensus recommendations for an integrated diagnostic approach to peripheral nerve sheath tumors arising in the setting of neurofibromatosis type 1
- Update on pediatric cancer surveillance recommendations for patients with neurofibromatosis type 1, Noonan syndrome, CBL syndrome, Costello syndrome, and related RASopathies
More Guidelines Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer