Summary
The cardiomyopathies are an important, heterogeneous group of heart muscle diseases that make a significant contribution to morbidity and mortality.[1] They are associated with mechanical and/or electrical dysfunction. Inappropriate ventricular hypertrophy or dilatation is usually present.[1] Cardiomyopathy involvement may be predominantly limited to the heart (primary cardiomyopathy) or form part of a generalized systemic disorder (secondary cardiomyopathy). Causes vary widely, but genetic etiologies are most common in primary cardiomyopathies. Complications include cardiovascular death and progressive heart failure, with its associated disability.[1]
Classifications
In 1995, the World Health Organization/International Society and Federation of Cardiology Task Force classified cardiomyopathies as primary myocardial disorders, whereas heart muscle diseases of known etiology or associated with systemic diseases were categorized as secondary or specific heart muscle diseases.[2] However, as research has improved the understanding of these conditions, working groups have proposed new but different classification systems.
The scientific statement from the American Heart Association has defined cardiomyopathies as "a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic. Cardiomyopathies either are confined to the heart or are part of generalized systemic disorders, often leading to cardiovascular death or progressive heart failure-related disability."[1]
Primary cardiomyopathies are those where the condition is predominantly confined to the heart muscle and where subclassifications of genetic, mixed, and acquired are adopted.
Secondary cardiomyopathies are those where myocardial involvement occurs as part of a systemic or multiorgan disorder.
The European Society of Cardiology Working Group on Myocardial and Pericardial Diseases has opted to use a clinical rather than genetic classification, where heart muscle disorders are classified according to morphology and function: "a myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease, hypertension, valvular disease and congenital heart disease sufficient to cause the observed myocardial abnormality."[3] Its members felt that many primary cardiomyopathies have significant extracardiac manifestations and that many secondary cardiomyopathies may involve the heart as the major manifestation.[Figure caption and citation for the preceding image starts]: Diseases that may cause cardiomyopathyElliott P, et al. Eur Heart J 2008; 29: 270-76; used with permission [Citation ends].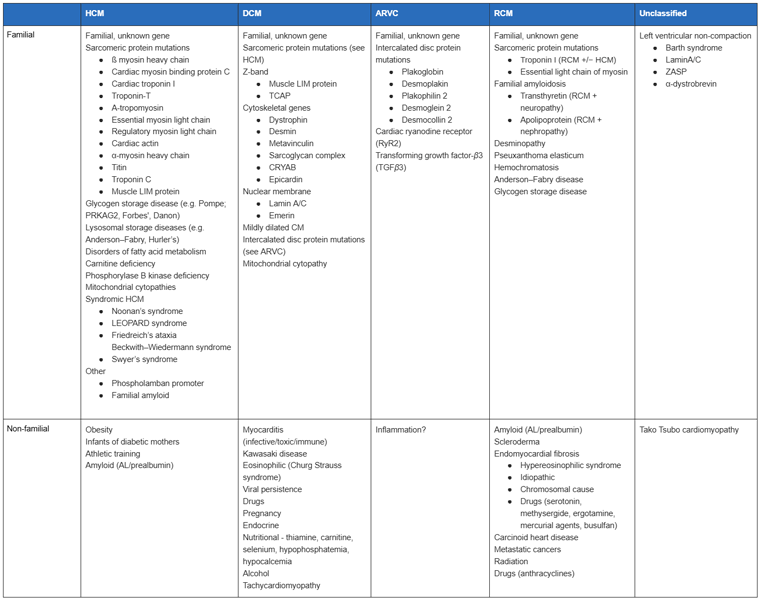
It is important to recognize that the traditional classification into hypertrophic, dilated, and restrictive cardiomyopathies mixes anatomical with functional designations, which are not mutually exclusive. [Figure caption and citation for the preceding image starts]: Proposed classification systemElliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270-276. Used with permission. [Citation ends].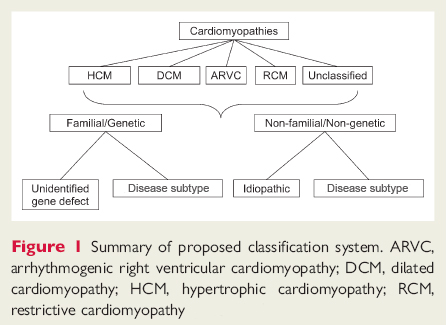
The American Heart Association published a separate scientific statement in 2019, covering the classification and diagnosis of cardiomyopathy in children.[4] This favours a classification for pediatric cases based primarily on the structural and functional phenotype (which forms the basis for diagnosis and management), with genetic and nongenetic causes as lower-level subcategories.
Differentials
Common
- Idiopathic dilated cardiomyopathy
- Myocarditis
- Alcohol: dilated cardiomyopathy
Uncommon
- Hypertrophic cardiomyopathy (HCM)
- Idiopathic restrictive cardiomyopathy
- Arrhythmogenic right ventricular cardiomyopathy (ARVC)
- Brugada syndrome and other ion channelopathies
- Conduction system disease
- Mitochondrial disorders
- Takotsubo syndrome
- Peripartum: dilated cardiomyopathy
- Tachycardia-induced: dilated cardiomyopathy
- Amyloidosis: hypertrophic or restrictive cardiomyopathy
- Hemochromatosis: restrictive or dilated cardiomyopathy
- Fabry disease: hypertrophic or restrictive cardiomyopathy
- Other lysosomal storage disease
- Doxorubicin: dilated cardiomyopathy
- Heavy metals/chemicals: dilated cardiomyopathy
- Diabetes mellitus
- Thyroid dysfunction: dilated cardiomyopathy
- Acromegaly: hypertrophic or dilated cardiomyopathy
- Noonan syndrome: hypertrophic cardiomyopathy
- Lentiginosis: hypertrophic cardiomyopathy
- Thiamine deficiency (wet beriberi): dilated cardiomyopathy
- Friedreich ataxia/muscular dystrophy: hypertrophic and dilated cardiomyopathy
- Deficiency of iron, niacin, selenium, or vitamin D: dilated cardiomyopathy
- Systemic lupus erythematosus: dilated cardiomyopathy
- Endomyocardial fibrosis/Loeffler endocarditis (hypereosinophilic syndrome)
- Sarcoidosis
- Electrolyte disorders
Contributors
Authors
Katie Linden, BMSc, MBChB
Cardiology Specialty Registrar
Belfast Health and Social Care Trust
Belfast
Northern Ireland
Disclosures
KL was reimbursed by Takeda UK Ltd., the manufacturer of Replagal for Fabry disease, for travel and accommodation for a leadership course. KL was reimbursed by Sanofi Aventis, the manufacturer of Genzyme for Fabry disease, for travel and accommodation for a conference. KL received a speaker fee from Sanofi Aventis, the manufacturer of Genzyme, for a talk on Fabry disease. There was no contractual right by Sanofi to control the content of this talk.
Alison Muir, MD
Consultant Cardiologist
Royal Victoria Hospital
Belfast Health and Social Care Trust
Belfast
UK
Disclosures
AM is an author of a reference cited in this topic.
Acknowledgements
KL and AM would like to gratefully acknowledge Dr Pascal McKeown, a previous contributor to this topic. PM is an author of a reference cited in this topic.
Peer reviewers
Lubna Choudhury, MB BCh, MD, MRCPI, FACC
Professor of Medicine
Division of Cardiology
Northwestern University
Chicago
IL
Disclosures
LC declares that she has no competing interests.
John Coltart, MD, FRCP, FACC, FESC, MRCS
Consultant Cardiologist
Cardio-thoracic Unit
Guy's and St Thomas' Hospital
London
UK
Disclosures
JC declares that he has no competing interests.
Vaikom S. Mahadevan, MBBS, MRCP
Consultant Cardiologist
Manchester Royal Infirmary
Manchester
UK
Disclosures
VSM declares that he has no competing interests.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.Full text Abstract
Ommen SR, Ho CY, Asif IM, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology joint committee on clinical practice guidelines. Circulation. 2024 Jun 4;149(23):e1239-311.Full text Abstract
Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.Full text Abstract
American College of Radiology. ACR appropriateness criteria: nonischemic myocardial disease with clinical manifestations. 2020 [internet publication].Full text
British Society of Echocardiography. Diagnosis and assessment of dilated cardiomyopathy: a guideline protocol from the British Society of Echocardiography. Jun 2017 [internet publication].Full text
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Patient information
Heart failure
More Patient informationVideos
Third heart sound gallop
Fourth heart sound gallop
More videos Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer