Summary
Definition
History and exam
Key diagnostic factors
- parent(s) diagnosed with sickle cell anemia, other sickle cell disease, or sickle cell trait
- persistent pain in skeleton, chest, and/or abdomen
- dactylitis
Other diagnostic factors
- high temperature
- pneumonia-like syndrome
- bone pain
- visual floaters
- tachypnea
- failure to thrive
- pallor
- jaundice
- tachycardia
- lethargy
- protuberant abdomen, often with umbilical hernia
- cardiac systolic flow murmur
- maxillary hypertrophy with overbite
- shock
Risk factors
- genetic
Diagnostic tests
1st tests to order
- DNA-based assays
- hemoglobin isoelectric focusing (Hb IEF)
- cellulose acetate electrophoresis
- high-performance liquid chromatography (HPLC) fractionation
- hemoglobin solubility testing
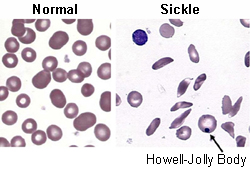
- peripheral blood smear
- CBC and reticulocyte count
- iron studies
Tests to consider
- pulse oximetry
- plain x-rays of long bones
- bacterial cultures
- CXR
Emerging tests
- noninvasive prenatal testing (NIPT)
Treatment algorithm
vaso-occlusive crisis
acute chest syndrome
chronic disease
Contributors
Authors
Sophie Lanzkron, MD, MHS
Director
Sickle Cell Center for Adults
Associate Professor of Medicine and Oncology
Johns Hopkins Medicine
Baltimore, MD
Disclosures
SL declares consultancy (Bluebird bio, Novo Nordisk, Pfizer, Novartis, Magenta); honoraria (Novartis); research funding (Imara, Novartis, GBT, Takeda, CSL-Behring, HRSA, PCORI, MD CHRC); stocks (Pfizer, Teva). SL is on the executive board of the National Alliance for Sickle Cell Centers (uncompensated).
Acknowledgements
Dr Sophie Lanzkron would like to gratefully acknowledge Dr Channing Paller, a previous contributor to this topic.
Disclosures
CP declares that she has no competing interests.
Peer reviewers
James Bradner, MD
Instructor in Medicine
Division of Hematologic Neoplasia
Dana-Farber Cancer Institute
Boston, MA
Disclosures
JB declares that he has no competing interests.
Adrian Stephens, MB BS, MD, FRCPath
Consultant Haematologist
University College London Hospitals
London
UK
Disclosures
AS declares that he has no competing interests.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
Bain BJ, Daniel Y, Henthorn J, et al. Significant haemoglobinopathies: a guideline for screening and diagnosis. Br J Haematol. 2023 Jun;201(6):1047-65.Full text Abstract
National Heart, Lung, and Blood Institute. Evidence-based management of sickle cell disease: expert panel report, 2014. Sep 2014 [internet publication].Full text
DeBaun MR, Jordan LC, King AA, et al. American Society of Hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv. 2020 Apr 28;4(8):1554-88.Full text Abstract
Chou ST, Alsawas M, Fasano RM, et al. American Society of Hematology 2020 guidelines for sickle cell disease: transfusion support. Blood Adv. 2020 Jan 28;4(2):327-55.Full text Abstract
Trompeter S, Massey E, Robinson S, et al. Position paper on International Collaboration for Transfusion Medicine (ICTM) Guideline 'Red blood cell specifications for patients with hemoglobinopathies: a systematic review and guideline'. Br J Haematol. 2020 May;189(3):424-27.Full text Abstract
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Differentials
- Gout
- Septic arthritis
- Connective tissue diseases
More DifferentialsGuidelines
- Guideline for the primary prevention of stroke
- Management of acute presentations of sickle cell disease
More GuidelinesPatient information
Sickle cell disease: what is it?
Sickle cell disease: what are the treatment options?
More Patient informationVideos
Venepuncture and phlebotomy: animated demonstration
Peripheral intravascular catheter: animated demonstration
More videos Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer