Summary
Definition
History and exam
Key diagnostic factors
- jugular venous distention
- lower extremity edema
- history of a chronic inflammatory condition, chronic infection, familial periodic fever syndrome
- history of monoclonal gammopathy of undetermined significance (MGUS)
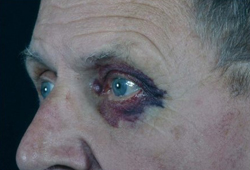
- periorbital purpura
- eyelid petechiae
- macroglossia
Other diagnostic factors
- fatigue
- unexplained weight loss
- dyspnea on exertion
- peripheral neuropathy
- autonomic neuropathy
- claudication
- nausea or vomiting
- abdominal cramps
- alternating bowel habit
- steatorrhea
- lightheaded
- submandibular salivary gland enlargement
- hepatomegaly
- shoulder pad sign
- diffuse muscular weakness
- orthostatic hypotension
- carpal tunnel syndrome
- musculoskeletal disorders
Risk factors
- monoclonal gammopathy of undetermined significance (MGUS)
- inflammatory polyarthropathy
- chronic infections
- inflammatory bowel disease
- familial periodic fever syndromes
- Castleman disease
Diagnostic tests
1st tests to order
- serum immunofixation electrophoresis
- urine immunofixation electrophoresis (using 24-hour urine collection)
- serum free light chain assay
- CBC with differential
- peripheral blood smear
- serum quantitative immunoglobulins
- serum protein electrophoresis
- comprehensive metabolic profile
- urine protein electrophoresis (using 24-hour urine collection)
- 24-hour total urine protein
- creatinine clearance
- orthostatic vital sign assessment
- tissue aspiration and biopsy
- fluorescence in situ hybridization (FISH)
Tests to consider
- mass spectrometry
- immunohistochemical studies
- immuno-electron microscopy
- genetic testing
- serum troponin T or I
- N-terminal pro-B-type natriuretic peptide (NT‐proBNP)
- lipid panel
- coagulation studies
- ECG
- echocardiogram (with tissue Doppler and global longitudinal strain)
- cardiac MRI (CMR)
- cardiac scintigraphy
- electromyogram/nerve conduction studies
- endocrine tests
- pulmonary function tests
- computed tomography (CT) scan
- fluorodeoxyglucose positron emission tomography (FDG-PET)/CT
- skeletal survey
- abdominal ultrasound
- gastric emptying scan
- upper and lower endoscopy
- 123I-labeled serum amyloid P (SAP) scintigraphy
- 6-minute walk test
Treatment algorithm
immunoglobulin light chain (AL) amyloidosis
AA amyloidosis (nonfamilial)
familial periodic fever syndromes
transthyretin (TTR) amyloidosis
refractory or relapsed AL amyloidosis
Contributors
Authors
Morie A. Gertz, MD, MACP

Seidler Jr. Professor of Medicine
Consultant in Hematology
Chair Emeritus of the Department of Medicine
Mayo Distinguished Clinician
Mayo Clinic College of Medicine
Rochester
MN
Disclosures
MAG has received personal fees from AbbVie and Arcellx for a Data Safety Monitoring board, personal fees from Ionis/Akcea, Prothena, Sanofi, and Janssen, and fees from Johnson & Johnson. MAG has received honoraria from Alnylam, AstraZeneca, Medscape, Dava Oncology, and Alexion. MAG is an author of several references cited in this topic.
Peer reviewers
Donna Reece, MD
Associate Professor of Medicine
Director
Program for Multiple Myeloma and Related Diseases
Princess Margaret Hospital
Toronto
Ontario
Canada
Disclosures
DR has been reimbursed by Millennium Pharmaceuticals, Inc and Johnson & Johnson, the manufacturers of bortezomib, for attending several conferences, for speaking at educational meetings, and for consulting work. She has also been reimbursed by Celgene, the manufacturer of lenalidomide and thalidomide, for attending several symposia and serving as a speaker.
Jeffrey Zonder, MD
Assistant Professor of Medicine and Oncology
Division of Hematology/Oncology
Wayne State University School of Medicine
Barbara Ann Karmanos Cancer Institute
Detroit
MI
Disclosures
JZ declares that he has no competing interests.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
Writing Committee, Kittleson MM, Ambardekar AV, et al. Transthyretin cardiac amyloidosis evaluation and management: 2025 ACC concise clinical guidance. J Am Coll Cardiol. 2026 Feb 10;87(5):549-65.Full text Abstract
National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis [internet publication].Full text
Muchtar E, Dispenzieri A, Gertz MA, et al. Treatment of AL amyloidosis: Mayo stratification of myeloma and risk-adapted therapy (mSMART) consensus statement 2020 update. Mayo Clin Proc. 2021 Jun;96(6):1546-77.Full text Abstract
Wechalekar AD, Cibeira MT, Gibbs SD, et al. Guidelines for non-transplant chemotherapy for treatment of systemic AL amyloidosis: EHA-ISA working group. Amyloid. 2023 Mar;30(1):3-17.Full text Abstract
Sanchorawala V, Boccadoro M, Gertz M, et al. Guidelines for high dose chemotherapy and stem cell transplantation for systemic AL amyloidosis: EHA-ISA working group guidelines. Amyloid. 2022 Mar;29(1):1-7. Abstract
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Differentials
- Hypertrophic cardiomyopathy (HCM)
- Membranous glomerulopathy
- Monoclonal gammopathy of undetermined significance (MGUS)-associated neuropathy
More DifferentialsGuidelines
- NCCN clinical practice guidelines in oncology: systemic light chain amyloidosis
- NCCN clinical practice guidelines in oncology: hematopoietic cell transplantation (HCT)
More Guidelines Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer