Summary
Definition
History and exam
Key diagnostic factors
- muscle strength fatigability
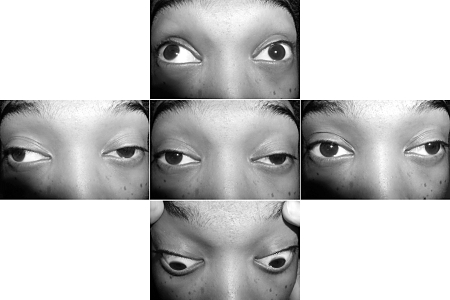
- ptosis
- diplopia
- dysphagia
- dysarthria
- facial paresis
- proximal limb weakness
- shortness of breath
Risk factors
- family history of autoimmune disorders
- genetic markers
- cancer-targeted therapy
Diagnostic tests
1st tests to order
- serum acetylcholine receptor (AChR) antibody analysis
- muscle-specific tyrosine kinase (MuSK) antibodies
- serial pulmonary function tests
Tests to consider
- striational receptor antibody assays
- repetitive nerve stimulation
- single-fiber EMG
- CT of chest
Treatment algorithm
myasthenic crisis
mild to moderate disease (class I to III)
severe (class IV or V) or refractory disease
Contributors
Authors
David P. Richman, MD
Distinguished Professor
Professor of Neurology
University of California - Davis
Davis
CA
Disclosures
DPR declares that he has no competing interests.
Acknowledgements
Dr David Richman would like to gratefully acknowledge Dr Robert Lisak, Dr Andrea Corse, and Dr Ami Mankodi, previous contributors to this topic.
Disclosures
AC and AM declare that they have no competing interests. RPL is a Data and Safety Monitoring Board Member for the COUR myasthenia gravis clinical trial. RPL is a site principal investigator for clinical trials and a co-author for the clinical trial reports for myasthenia gravis therapies for Alexion, Argenx, and UCB Ra. RPL's institution receives payment for the time spent on these clinical trials. RPL has received book royalties from Oxford University Press and Blackstone, and has carried out consultancy work for Avilar.
Peer reviewers
Vern C. Juel, MD
Associate Professor of Medicine (Neurology)
Duke University
Durham
NC
Disclosures
VCJ declares that he has no competing interests.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
Narayanaswami P, Sanders DB, Wolfe G, et al. International consensus guidance for management of myasthenia gravis: 2020 update. Neurology. 2021 Jan 19;96(3):114-22.Full text Abstract
Skeie GO, Apostolski S, Evoli A, et al. Guidelines for treatment of autoimmune neuromuscular transmission disorders. Eur J Neurol. 2010 Jul;17(7):893-902.Full text Abstract
Gronseth GS, Barohn R, Narayanaswami P. Practice advisory: thymectomy for myasthenia gravis (practice parameter update). Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2020 Apr 21;94(16):705-9.Full text Abstract
Elovaara I, Apostolski S, van Doorn P, et al. EFNS guidelines for the use of intravenous immunoglobulin in treatment of neurological diseases: EFNS task force on the use of intravenous immunoglobulin in treatment of neurological diseases. Eur J Neurol. 2008 Sep;15(9):893-908.Full text Abstract
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Differentials
- Lambert-Eaton myasthenic syndrome (LEMS)
- Botulism
- Penicillamine-induced myasthenia gravis
More DifferentialsGuidelines
- International consensus guidance for management of myasthenia gravis: 2020 update
- Practice advisory: thymectomy for myasthenia gravis (practice parameter update)
More GuidelinesPatient information
Myasthenia gravis
More Patient informationVideos
Venepuncture and phlebotomy: animated demonstration
Peripheral intravascular catheter: animated demonstration
More videos Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer