Summary
Definition
History and exam
Key diagnostic factors
- family history of autosomal-dominant PKD (ADPKD) or end-stage renal disease
- family history of cerebrovascular event
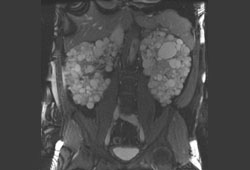
- renal cysts
- hypertension
- abdominal/flank pain
- hematuria
- palpable kidneys/abdominal mass
- headaches
- dysuria, suprapubic pain, fever
Other diagnostic factors
- cardiac murmur
- abdominal hernia
- hepatomegaly
- chest pain
Risk factors
- family history of autosomal-dominant PKD (ADPKD)
- family history of cerebrovascular event
Diagnostic tests
1st tests to order
- renal ultrasound
- CT abdomen/pelvis
- MRI abdomen/pelvis
- urinalysis/Gram stain and urine culture
- serum electrolytes, BUN, creatinine
- fasting lipid profile
- ECG
- CT brain
Tests to consider
- genetic testing
- echocardiogram
- 24-hour urine collection
- dual-energy CT of abdomen/pelvis
- lumbar puncture and cerebrospinal fluid analysis
- C-reactive protein
- PET scan
Treatment algorithm
confirmed autosomal-dominant polycystic disease
end-stage renal disease
Contributors
Authors
Marie C. Hogan, MD, PhD
Consultant
Division of Nephrology
Professor of Medicine
College of Medicine
Mayo Clinic
Rochester
MN
Disclosures
MCH receives research funding from Novartis, and was an investigator participating in tolvaptan clinical trials. MCH is also an author of several references cited in this topic.
Maria Irazabel Mira, MD, PhD
Associate Professor of Medicine, Associate Consultant
Division of Nephrology and Hypertension
Department of Internal Medicine
Mayo Clinic
Rochester
MN
Disclosures
MIM is an author of several references cited in this topic.
Acknowledgements
Dr Marie C Hogan and Dr Maria Irazabal Mira would like to gratefully acknowledge Dr Vicente Torres, a previous contributor to this topic.
Disclosures: VT is an author of several references cited in this topic.
Peer reviewers
Richard Sandford, PhD, FRCP
Wellcome Trust Senior Fellow in Clinical Research
University Reader in Real Genetics
Honorary Consultant in Medical Genetics
Cambridge
UK
Disclosures
RS declares that he has no competing interests.
Arlene Chapman, MD
Professor of Medicine
Renal Division
Emory University
School of Medicine
Atlanta
GA
Disclosures
Not disclosed.
Peer reviewer acknowledgements
BMJ Best Practice topics are updated on a rolling basis in line with developments in evidence and guidance. The peer reviewers listed here have reviewed the content at least once during the history of the topic.
Disclosures
Peer reviewer affiliations and disclosures pertain to the time of the review.
References
Key articles
Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018 Dec 6;4(1):50.Full text Abstract
Cornec-Le Gall E, Alam A, Perrone RD. Autosomal dominant polycystic kidney disease. Lancet. 2019 Mar 2;393(10174):919-35. Abstract
Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009 Jan;20(1):205-12.Full text Abstract
Torres VE, Chapman AB, Devuyst O, et al; TEMPO 3:4 Trial Investigators. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012 Dec 20;367(25):2407-18.Full text Abstract
Schrier RW, Abebe KZ, Perrone RD, et al; HALT-PKD Trial Investigators. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014 Dec 11;371(24):2255-66. Abstract
Torres VE, Abebe KZ, Chapman AB, et al; HALT-PKD Trial Investigators. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N Engl J Med. 2014 Dec 11;371(24):2267-76. Abstract
Reference articles
A full list of sources referenced in this topic is available to users with access to all of BMJ Best Practice.
Differentials
- Acquired cystic kidney disease
- Simple cyst
- Tuberous sclerosis complex
More DifferentialsGuidelines
- Autosomal-dominant polycystic kidney disease (ADPKD): executive summary
More GuidelinesCalculators
Glomerular Filtration Rate Estimate by the IDMS-Traceable MDRD Study Equation
More CalculatorsVideos
Diagnostic lumbar puncture in adults: animated demonstration
More videos Log in or subscribe to access all of BMJ Best Practice
Log in or subscribe to access all of BMJ Best Practice
Use of this content is subject to our disclaimer